BoltzGen: from designs to data in 2 weeks
BoltzGen: from designs to data in 2 weeks
/// NEED TO FINISH INTRO AND ADD IN HANNES’ REPLIES ///
We believe the future of protein engineering lies in low-latency design-build-test cycles. Recently, the Boltz team reached out with an ambitious goals: to validate BoltzGen, their recent all-atom generative model capable of generating binders to almost any target, and we knew this was the right opportunity to demonstrate the Adaptyv thesis.
Universal binder design with BoltzGen
BoltzGen is a general-purpose all-atom generative model trained to design binders against any kind of biomolecular target, including proteins, peptides, RNA, DNA, or even small molecules. It builds on the Boltz-2 architecture and a diffusion training process, yet adds some novel elements compared to the former and other key players in the binder design field.
We found
encoding
training
selection and validation
challenging set of targets
For example, instead of treating sequence and structure as separate problems, it learns both at once. The same diffusion process that predicts a structure can also design a new sequence to fit it.
A key innovation is its geometric encoding of residue identity. Instead of discrete amino acid tokens, BoltzGen encodes residues through their all-atom geometry, allowing continuous diffusion while still inferring residue types from structure. This lets the model reason about binding interfaces in full atomic detail and design directly in 3D.
BoltzGen also introduces a design specification language, letting users specify covalent bonds, binding sites, partial scaffolds, or flexibility constraints. It can generate cyclic peptides, disulfide-bonded loops, or nanobody CDRs within a single unified interface.
In short: one model for all binder types, controlled by flexible design constraints.
BoltzGen stands out as one of the first truly general-purpose models for biomolecular design. It doesn’t just predict protein structures — it designs new molecules, atom by atom, capable of binding to proteins, peptides, RNA, DNA, or small molecules. What makes it different is how it merges the physical and generative worlds into a single system.
1. All-atom encoding
Most design models work at the residue or backbone level. BoltzGen instead learns directly from all atoms.
Each residue is represented geometrically rather than symbolically — meaning the model doesn’t just guess the amino acid letter, it infers it from the atomic layout. This continuous encoding lets the diffusion process “think” in three-dimensional space, capturing the subtle geometry of interactions like hydrogen bonds, salt bridges, or steric packing.
This design removes the gap between discrete protein sequence models and continuous structure predictors. BoltzGen can fold, design, and reason about binding in the same atomic frame of reference.
2. Unified training across structure prediction and design
BoltzGen is trained on millions of experimental and self-distilled structures, but unlike previous models, it doesn’t separate folding from design.
During training, the model randomly decides which parts of each structure to predict and which to redesign. This forces it to learn both how proteins fold and how different molecules interact — from proteins to nucleic acids and small ligands.
This multi-task approach means a single model can scaffold motifs, generate nanobody CDRs, design cyclic peptides, or predict complexes, all without retraining. The same learned physics applies to every modality.
3. Selection and experimental validation
BoltzGen’s pipeline doesn’t stop at generation. After producing tens of thousands of candidates per target, it passes each through a filtering and ranking process combining physics-based and learned metrics — such as interface area, hydrogen bonds, and Boltz-2 confidence scores.
At Adaptyv, we connected directly to this pipeline. Once the top-ranked designs were selected, sequences were sent to our automated platform for expression and biophysical validation. Each campaign — from digital design to affinity measurement — was completed in under two weeks.
This tight loop between AI generation and automated wet-lab validation made it possible to test large numbers of completely novel designs faster than ever before.
4. A deliberately hard target set
To measure true generalization, the Boltz and MIT teams chose nine protein targets that had no bound structures anywhere in the PDB and less than 30% sequence identity to any known complexes. These are the kinds of proteins for which no model has seen examples of binding.
The results were striking: with just 15 designs per target, BoltzGen produced nanomolar binders for six of nine of these completely unseen proteins. This level of success on hard, low-similarity targets suggests the model is not memorizing — it’s learning the underlying physics of molecular binding.
Together, these ingredients make BoltzGen a milestone for AI-driven protein design.
It doesn’t just create new sequences; it understands how atoms meet, fold, and interact.
Combined with Adaptyv’s rapid validation, this makes for a closed discovery loop — from digital diffusion to measured binding data in less than two weeks.
1. Design specification language
BoltzGen introduces a flexible “design specification” interface — essentially a promptable grammar for molecular control. You can define which residues should bind, remain flexible, or be fixed; constrain distances; specify covalent bonds (for cyclic or disulfide-stapled peptides); or even provide partial structural motifs.
This gives researchers programmable control over what the model generates, something no previous general binder design system offered in such a unified way.
4. Integrated inverse folding and ranking pipeline
The BoltzGen pipeline goes beyond the generative model. It adds a series of steps — inverse folding, refolding with Boltz-2, and physics-based scoring — to automatically select designs that are stable, soluble, and likely to express.
This helps bridge the gap between purely computational design and real-world expression success, enabling smoother transfer to automated labs like Adaptyv.
7. End-to-end open release
The full codebase, model weights, training data, and inference tools are all released under the MIT license.
This open access is rare for a model of this scale and complexity, effectively turning BoltzGen into a public platform for anyone to test binder design pipelines or benchmark against it.
Beyond its core architecture and validation, BoltzGen pushes the field forward in other ways. It comes with a programmable design language, letting researchers specify structural motifs, covalent bonds, or flexible regions directly in the generation prompt. Its
continuous all-atom diffusion
captures both chemistry and geometry at once, producing realistic interactions rather than idealized folds.
BoltzGen also unifies design with inverse folding, ranking, and filtering, automatically surfacing soluble, stable candidates. Finally, the model’s
target diversity metrics
show it doesn’t just reuse known motifs — it truly learns new binding solutions.
Together, these innovations make BoltzGen one of the most complete and transparent frameworks for universal binder design to date.
Results from our Adaptyv Foundry - screenshots from Proteinbase
Mini-binders
We can show results from Proteinbase
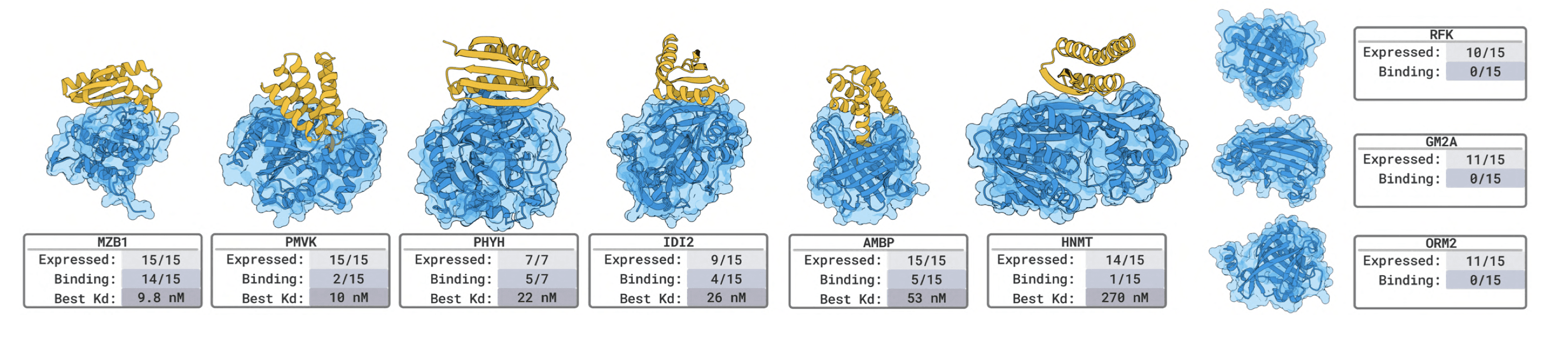
Novel targets hit-rates
Standard targets hit-rates
Nanobodies
We can show results from Proteinbase
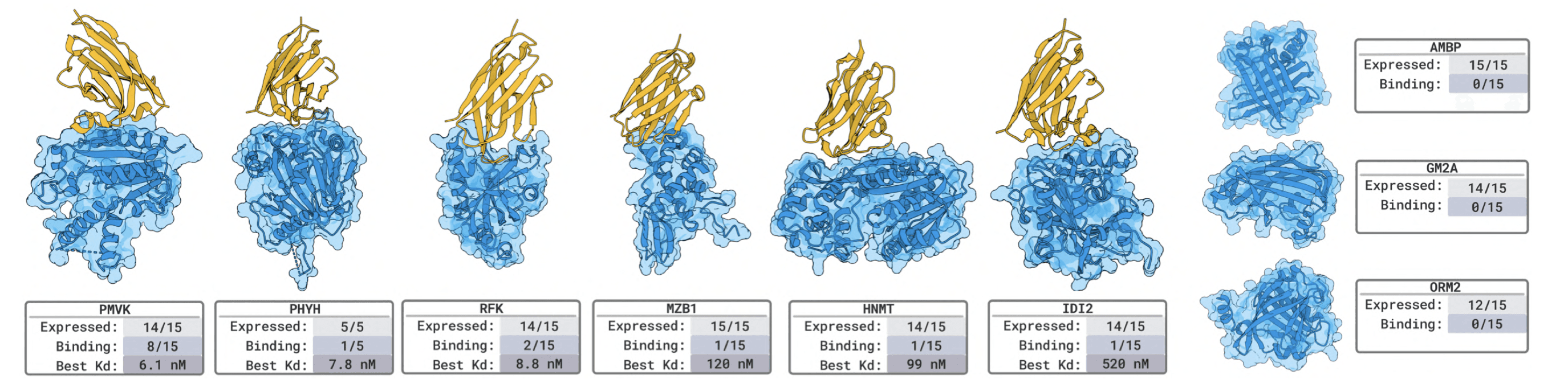
Novel targets hit-rates
Standard targets hit-rates
How we did all these experiments in <2 weeks - workflow overview and more insights
All validation was performed on our automated cell-free expression and binding affinity measuring platform. This involves:
We received digital sequences directly from the Boltz team and ran expression and binding assays at scale without manual intervention.
Each batch followed the same flow:
Design intake and sequence processing
Automated DNA synthesis and in vitro translation
Affinity testing via BLI and SPR
Data upload and aggregation
Because all steps are integrated, we were able to generate and validate hundreds of designs in parallel, turning BoltzGen’s digital predictions into experimental data in under 10 working days.
This kind of closed-loop validation is what makes AI-native protein design feasible at scale — rapid iteration between computational generation and experimental verification.
Find all the data on Proteinbase - links
All results, sequences, expression data, and affinity measurements are now available on Proteinbase.
You can explore BoltzGen’s designs, filter by target, and compare affinities across nanobody and miniprotein modalities.
The dataset is fully open, forming one of the largest experimental benchmarks for all-atom binder design to date.
QnA with Hannes - will add
Waiting for his reply…
1. What does a “universal binder design model” mean?
2. Why design a universal model instead of focusing on one modality like nanobodies?
3. What’s novel in BoltzGen and how did you come to/ think about these approaches?
4. BoltzGen is open-sourced under an MIT license. Why did you decide to do this in quite a competitive protein design model-building market (e.g., Chai, Latent, DeepMind)
5. You have a pretty sizeable and impressive team behind BoltzGen. How did it feel to manage/collaborate with this team?
6. Your target selection is quite unique. Why did you choose the exact targets you validated with us and how did you select them?
7. Why did you go with Adaptyv for validating your binders?
8. What did Adaptyv make possible in this project? How was your experience?
9. What’s next for BoltzGen and what sort of experiments are you interested in conducting?
10. What should we expect from the Boltz team in the future?
11. What protein design and data initiative would you like to see the community (and Adaptyv) do?
What does a “universal binder design model” mean?The three main points the universality pertains to are:
The designed binders can be nanobodies, (cylic, disulfide-bonded)-peptides, or any other type of protein.
The target can be any biomolecule (DNA, RNA, small molecules, proteins with or without PTMs, …).
Controllability and design constraints: BoltzGen comes with an expressive design specification language over covalent bonds, structure constraints, binding sites, and more.
2. Why design a universal model instead of focusing on one modality like nanobodies?As models learn to emulate physics primarily through examples provided, we believe expanding the generality of the method further improves its design capabilities for specific classes as well. The biomolecular interactions in different classes are based on the same underlying physics and these are the well-generalizing patterns that allow for discovering designs against novel targets.3. What’s novel in BoltzGen and how did you come to/ think about these approaches?The three aspects I would point out are:
We think differently about binder design model development: strong folding should be the optimization goal. It is hard to have one model do both as evidenced by BoltzGen being the first to match SOTA folding performance, but design requires strong reasoning about target-binder interactions - precisely what the folding task teaches a model.
Our design specification language:
Try it yourself
BoltzGen is open source at github.com/HannesStark/boltzgen.
All wet-lab results are on Proteinbase.
Adaptyv Bio — test your proteins.


