Nov 5, 2025
Designer Spotlight: Navigating the multi-property maze for therapeutic peptide design - Tong Chen
Tong Chen and colleagues from Pranam Chatterjee’s Programmable Biology Group at the University of Pennsylvania are showcasing their most recent model MOG-DFM - a discrete flow matching model capable of optimizing therapeutic peptides across multiple (up to 5) different properties

Introducing designed peptides
You’ve probably heard about GLP-1 peptides: they’re the technology behind drugs like Ozempic and Wegovy that have transformed diabetes and obesity treatment. But what you might not know is that it took decades of work to make these peptides be clinically viable, meaning that they not only bind their targets effectively, but also survive in the body, avoid toxicity, dissolve well, and actually work as safe medicines. Most peptide candidates fail somewhere along this pipeline, not because they can’t bind, but because they don’t have a combination of good therapeutic properties. Now imagine if we could design GLP-1-like peptides for any disease and have them be therapeutically viable from the very beginning.
Recent advances in machine learning are making this vision possible. Instead of tweaking one molecule at a time, researchers now use generative models (basically algorithms that can explore vast sequence spaces) to imagine entirely new peptide candidates. The Programmable Biology Group has pushed the boundaries with models for motif-specific targeting (moPPIt), fusion-breakpoint detection (SOAPIA, FusOn-pLM), post-translational modification prediction (PTM-Mamba), the design of non-canonical or cyclized peptides (PepTune), and target-binding peptides for rare-disease treatment (with the recent Gumbel-Softmax flow matching framework).
However, the development of therapeutic peptides involves more than just designing a peptide that binds effectively to its target. Researchers must consider a variety of physical and biological properties, such as solubility, stability, affinity, hemolysis, and non-fouling behavior. The challenge lies in balancing these conflicting properties, as optimizing one may negatively affect another.
In this post, we’ll break down the group’s newest model, Multi-Objective-Guided Discrete Flow Matching (MOG-DFM), which addresses these challenges by leveraging a novel multi-objective optimization algorithm for discrete flow matching to guide the generation of peptide sequences that optimize multiple properties at the same time. We have validated their designs for two properties we can readily test: binding and expression, with some impressive results!

Why is peptide design such a challenging problem?
Designing peptides that satisfy multiple, often conflicting, functional and biophysical criteria is no simple task. The difficulty arises on two intertwined fronts. First, unlike single-objective problems where there is a clear optimum, multi-objective optimization (MOO) yields a Pareto front of trade-off solutions where improving one property typically degrades another. The second difficulty is biological: the properties themselves are not independent and the mapping from sequence to phenotype is nonlinear, due to epistatic interactions and context-dependent effects.
MOG-DFM for a more efficient multi-objective optimization
MOG-DFM offers a novel solution to this problem by integrating both generative sequence modeling with multi-objective optimization. At is core, MOG-DFM leverages discrete flow matching (DFM), a generative modeling approach tailored for biological sequence data like peptides. DFM learns how a sequence should evolve, step by step, from a random initialization toward a realistic, functional target. This involves the concept of a “velocity field”: for each position in the sequence, the model predicts the probability (or “velocity”) of switching the current token (such as an amino acid) to any other possible choice. Unlike methods that operate in continuous space, DFM works natively with discrete symbols, making it especially suitable for applications in biology.
To design a new peptide, MOG-DFM begins with a random sequence and sets a specific “trade-off direction” that represents the desired balance among properties (such as affinity vs. solubility) from a Das–Dennis simplex lattice . This direction is chosen so that different runs of the algorithm can explore different parts of the possible trade-off landscape.
After initialization, MOG-DFM will perform multiple sampling steps to gradually evolve the starting sequences to ones with desired properties. At each step, MOG-DFM selects one random position in the sequence to update. For that position, the algorithm evaluates all possible candidate tokens by calculating two scores: a rank score, reflecting how much each option improves the desired properties compared to the alternatives, and a directional score, which measures how well the change moves the sequence toward the chosen trade-off direction. These are combined into a guidance score, which adjusts the underlying DFM “velocity” for each possible transition. In practice, transitions with higher guidance scores are exponentially favoured, actively steering sequence evolution toward peptides that optimally balance all objectives.
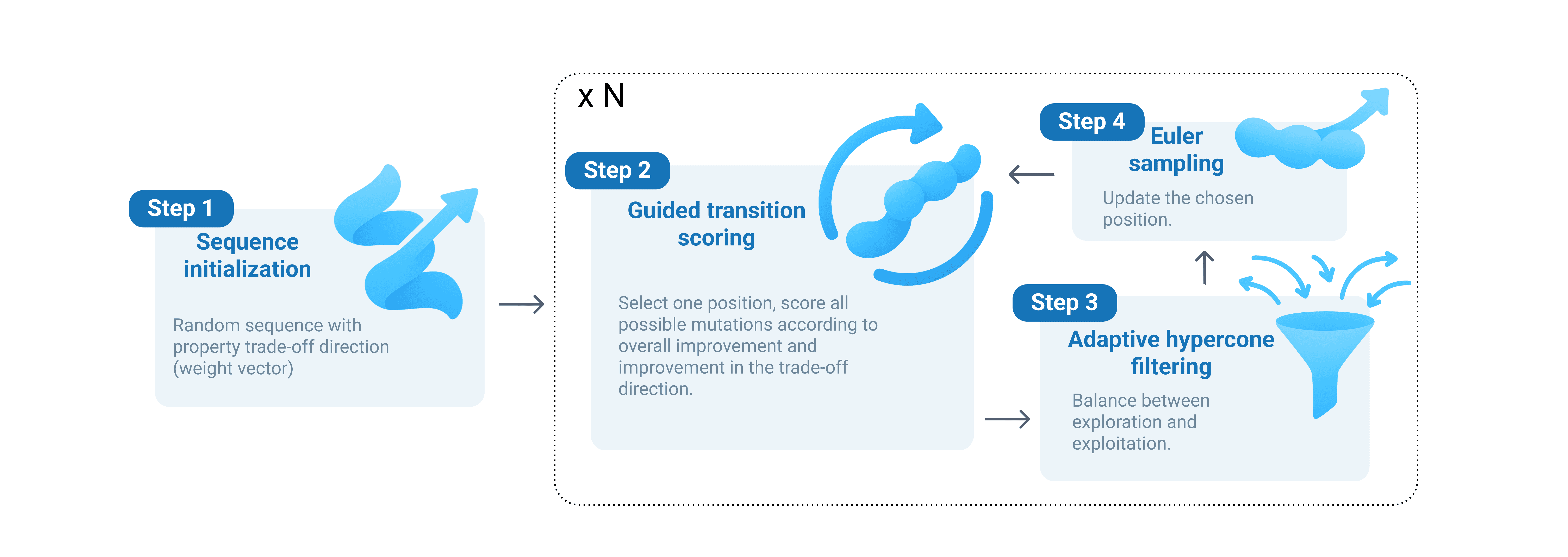
To keep the generative process efficient, MOG-DFM uses a technique called adaptive hypercone filtering. Imagine each possible sequence change as an arrow pointing in a direction: some arrows point toward the desired trade-off (the optimal balance of properties), while others don’t. Hypercone filtering works by only allowing changes whose arrows fall within a certain angle (“cone”) of the target direction. If the algorithm finds itself with too few options, the cone automatically widens, encouraging more exploration! If it’s admitting too many, the cone narrows to maintain focus. This dynamic adjustment helps the algorithm avoid both getting stuck and wandering aimlessly. The sequence is then updated using the Euler method: the chosen position is switched to the selected token with a probability determined by its transition velocity to the selected token, otherwise the original token is retained. This approach maintains stochasticity in the sequence evolution, while ensuring that updates remain consistent with the desired trade-off direction.
Overall, discrete flow matching supplies the underlying generative backbone, while MOG-DFM injects multi-objective guidance through the rank-directional scores and maintains exploitation and exploration balance via the adaptive hypercone filtering mechanism.
In silico MOG-DFM benchmarks
MOG-DFM was benchmarked on a peptide binder design task guided simultaneously by five therapeutic properties:
Hemolysis: A measure of toxicity, specifically the ability of a peptide to damage red blood cells. Lower values indicate safer, less toxic peptides.
Non-fouling: Reflects the peptide’s tendency to avoid sticking to unintended surfaces, reducing unwanted interactions and side effects.
Solubility: Determines how readily the peptide dissolves in biological fluids, a key factor for delivery and bioavailability.
Half-life: Indicates the stability of the peptide in the body. Longer half-life means the peptide persists longer, allowing for lower dosing and improved efficacy.
Binding affinity: Measures how tightly the peptide binds to its intended target, such as a disease-related protein or receptor.
Benchmarking was performed using a set of protein targets that included structured proteins with pre-existing binders, structured proteins without known binders, and intrinsically disordered proteins. Significantly, MOG-DFM-designed peptides consistently achieve low hemolysis (0.06–0.09), high non-fouling (>0.78) and solubility (>0.74), extended half-life (28–47 h), and good affinity scores (6.4–7.6).

MOG-DFM was also compared to four classical multi-objective optimization baselines: NSGA-III, SMS-EMOA, SPEA2, and MOPSO. Although MOG-DFM incurs longer runtimes, it consistently yields superior trade-offs: it reduces predicted hemolysis by over 10%, increases non-fouling and solubility by roughly 30–50%, and extends half-life by a factor of three to four relative to the next-best competitor, while maintaining comparable affinity. These results highlight MOG-DFM’s ability to navigate high-dimensional, conflicting property landscapes and produce peptide binders with well-balanced profiles that would be difficult to obtain via traditional optimizers.
Target | Method | Time (s) | Hemolysis (↓) | Non-Fouling | Solubility | Half-Life | Affinity |
|---|---|---|---|---|---|---|---|
1B8Q | MOPSO | 8.54 | 0.1066 | 0.4763 | 0.4684 | 4.449 | 6.0594 |
NSGA-III | 33.13 | 0.0862 | 0.5715 | 0.5825 | 7.324 | 7.2178 | |
SMS-EMOA | 8.21 | 0.1196 | 0.3450 | 0.3511 | 3.023 | 5.955 | |
SPEA2 | 17.48 | 0.0819 | 0.4973 | 0.5057 | 4.126 | 7.324 | |
MOG-DFM | 43.00 | 0.0785 | 0.8445 | 0.8455 | 27.227 | 5.9094 | |
PPP5 | MOPSO | 11.34 | 0.0883 | 0.4711 | 0.4255 | 1.769 | 6.6958 |
NSGA-III | 37.30 | 0.0479 | 0.7138 | 0.7066 | 2.901 | 7.3789 | |
SMS-EMOA | 8.43 | 0.1242 | 0.4269 | 0.4334 | 1.031 | 6.2854 | |
SPEA2 | 19.02 | 0.0555 | 0.6221 | 0.6098 | 2.613 | 7.6253 | |
MOG-DFM | 90.00 | 0.0617 | 0.7738 | 0.751 | 27.775 | 6.8197 |
Benchmarking MOG-DFM and 4 other multi-objective optimization baseline on all 5 key properties for therapeutic peptides. Table provided by the Programmable Biology Group.
Validating 24 MOG-DFM designs in our Adaptyv Foundry
We received 24 ten-residue de novo peptide binders targeting human FcRn, We then experimentally characterized all 24 designs by Biolayer Interferometry (BLI) using our Affinity Characterization workflow, with two replicates per peptide. We can see 6/24 sequences with clear KDs in the hundreds of nanomolar range (defined as binders by our assay, but still with some room for optimization).
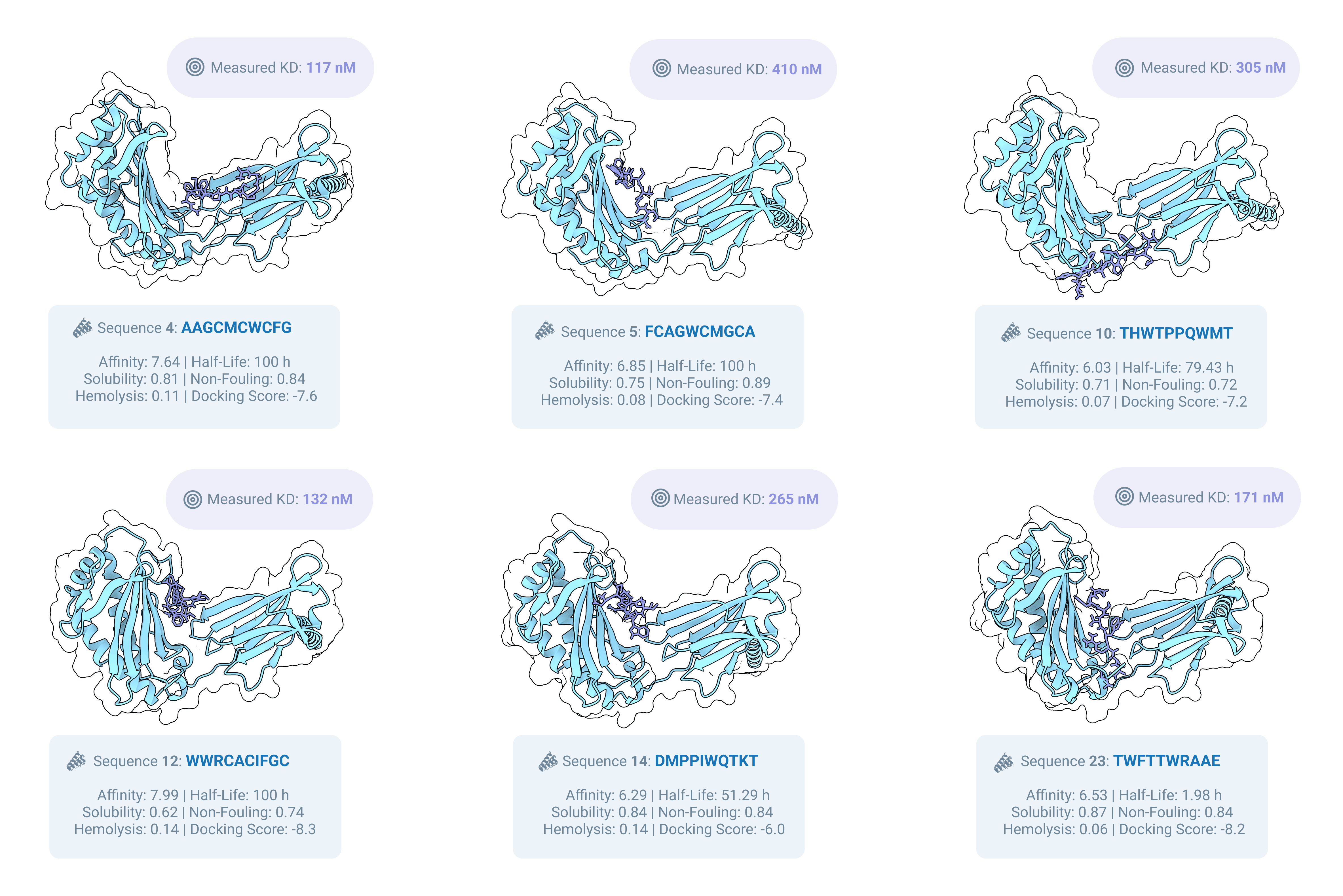
What’s next for MOG-DFM
As noted before, the framework can become computationally intensive as sequence length or output dimensionality grows. Extending to longer proteins or other high-dimensional biological sequences will increase the number of candidate transitions per step and and the number of sampling iterations needed. Second, while MOG-DFM steers generation toward Pareto-efficient regions, it does not come with theoretical guarantees of Pareto optimality or coverage. The adaptive guidance and hypercone filtering induce positive expected improvement in the desired directions, but there is no formal assurance that the sampled set will fully represent or converge to the true Pareto front.
Thus, these limitation motivate two directions to improve upon:
1. Scale MOG-DFM to longer sequences, including those with non-canonical amino acids,
2. Strengthen Pareto convergence guarantees and better characterizing coverage, potentially via uncertainty-aware or feedback-driven extensions to the guidance mechanism.
Resources and links
The MOG-DFM experimental results are hosted on Proteinbase.
Check out what the Programmable Biology Group is working on!
We thank both Tong Chen and Prof. Pranam Chatterjee for actively working on this blog post!
Have some novel proteins you want to test in the lab? Come talk to us — we’d like to run many more of those protein designer spotlights, so if you have a cool new hypothesis or model to test we’d love to hear from you!


